If you or someone you love is dealing with brain fog, chronic headaches, anxiety, depression, anhedonia, memory problems, sleep issues, word finding difficulty or has been told their neurological symptoms are “stress” or “just getting older” — this article is for you. It is long and detailed, because the truth is detailed.
THE SHORT VERSION
Your brain is not inflamed for no reason. Neuroinflammation— inflammation in the brain— is often what is behind brain fog, many headaches, mood changes, and cognitive decline. It has causes, and those causes can often be identified and addressed.
It rarely starts in the brain. It often starts at the level of barriers and immune tolerance—especially the gut, where much of your immune system interfaces with the outside world. When the gut barrier breaks down, the immune system loses calibration. Inflammation spreads. The barrier protecting the brain becomes more vulnerable too—and now the brain is exposed to inflammatory signals, toxicants, infections, and an immune system that has lost its way.
Many contributors can stack: gut dysbiosis and opportunistic overgrowth, mold exposure and mycotoxins (including via the nasal/olfactory route), heavy metals that disrupt mineral-dependent brain function, chronic infections and viral reactivation when immune regulation fails, pesticide exposures associated with injury to vulnerable dopaminergic neurons, and even old head injuries that can “prime” the brain to overreact to future insults.
Inflammation also affects blood flow. Chronic inflammatory signaling can activate coagulation and reduce microvascular perfusion. Neurons that aren’t getting enough oxygen can’t think, signal, or heal properly—this alone can create the “foggy” feeling many clients describe.
And once barriers are compromised, the immune system can begin targeting neural tissue. Autoantibodies are one way this shows up in complex cases: a signature of lost tolerance. Meanwhile, mitochondria shift from energy production into danger signaling—what Dr. Naviaux describes as the Cell Danger Response—locking the system into fatigue, sensitivity, and poor resilience.
The goal is not simply suppressing inflammation—it’s restoring regulation: remove the burdens, heal the barriers, and rebuild immune balance.
What follows is the full, detailed exploration of each of these mechanisms and how they interconnect. If you are a practitioner, a researcher, or someone who wants to understand the complete picture, read on.
A note from the author: The perspectives shared in this article reflect my clinical experience working with complex, multi-system clients. I am not stating absolutes — I am sharing the patterns, mechanisms, and truths I have observed in practice, informed by the published literature and by the real people I have had the privilege of working with. I am ever evolving. As the field advances, as new research emerges, and as new insights reshape our understanding, I will update this work accordingly. Science is not static, and neither am I. If you are reading this and something has changed since it was written, know that I am likely already integrating it into my practice and my thinking.
Educational information only; not every mechanism applies to every person. Seek medical care for new or severe neurological symptoms.
IN THIS ARTICLE
1. The Gut: Where Tolerance Is Made and Lost
2. The Oral Microbiome: An Amplifier That Can Become a Primary Driver
3. Blood-Brain Barrier Permeability: The Convergence Point
4. Stealth Infections: When the Immune System Cannot Keep Up
5. Toxic Metals: Displacing What the Brain Needs Most
6. Copper, Mineral Dysregulation, and Neuroexcitotoxicity
7. EMF Exposure and Voltage-Gated Calcium Channels
8. Biotoxins and the Olfactory Nerve: A Direct Route to the Brain
9. PANS: A Window into Immune-Mediated Neuroinflammation
10. Traumatic Brain Injury: The Priming Event
11. COVID-19: The Straw That Broke Immune Tolerance
12. Microglial Activation: Where Immune and Nervous Systems Become One
13. Mast Cell Activation, Histamine, and Sleep Disruption
14. Hypercoagulation and Neuronal Hypoxia: The Silent Starve
15. Insulin Resistance: Metabolic Fire in the Brain
16. Hormonal Dysregulation and Autoimmune Encephalopathy
17. Membrane Integrity: Plasmalogens and Phosphatidylcholine
18. Mitochondrial Dysfunction and the Cell Danger Response
19. Methylation: The Quiet Bottleneck
20. Immune Regulation: The Thread That Ties It All Together
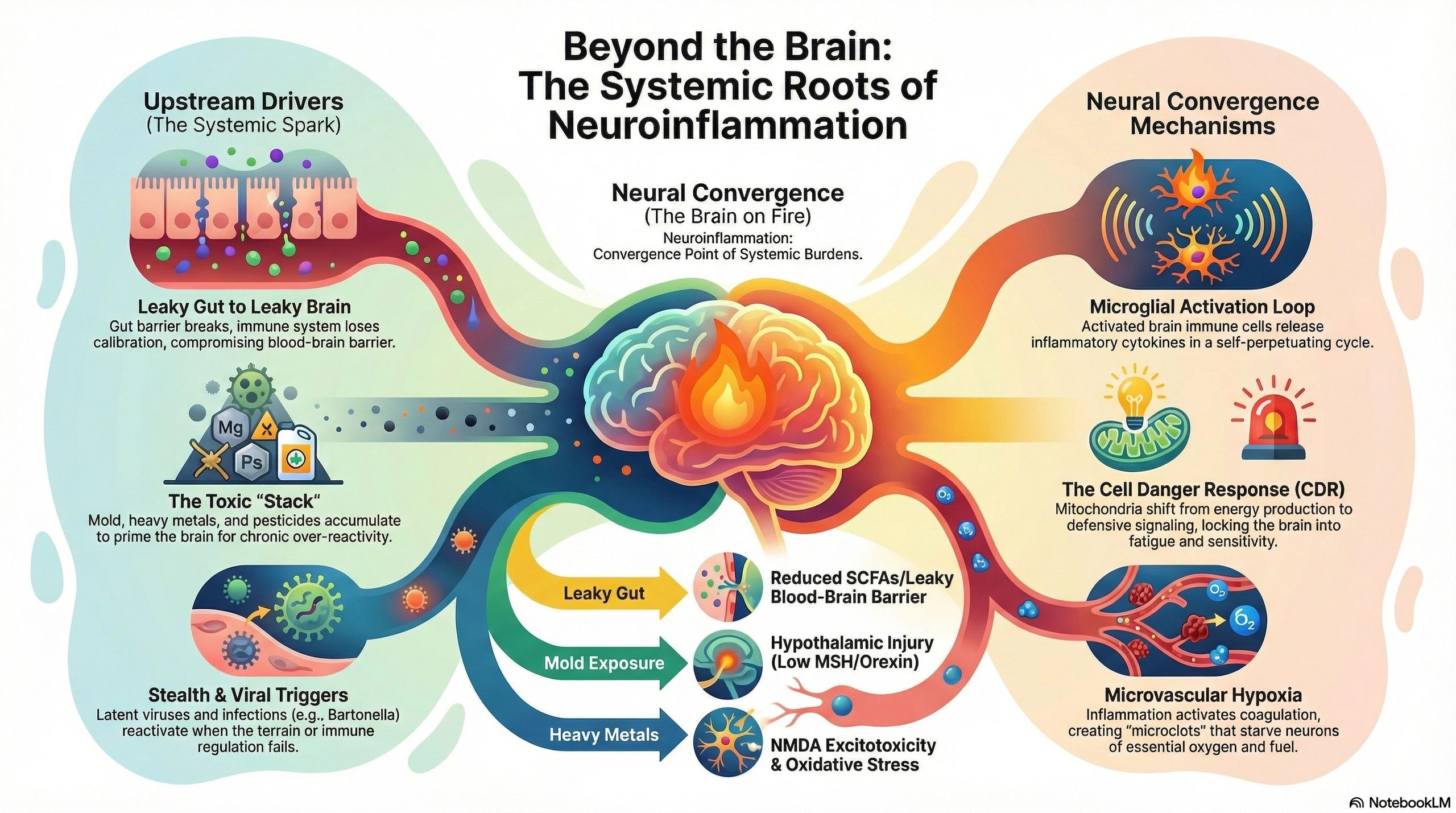
Neuroinflammation is not a diagnosis — it is a convergence point. It is the final common pathway through which a wide array of upstream insults — infections, toxins, metabolic dysfunction, immune dysregulation — express themselves in the central nervous system. When clients present with brain fog, mood instability, autonomic dysfunction, sleep disruption, and cognitive decline, the question is rarely simply whether neuroinflammation is present. The question is what lit the fire, and what is keeping it burning.
In clinical practice, I rarely see a single cause. On advanced neural autoantibody panels, I consistently see autoantibodies to brain and nervous system tissue that reflect the cumulative burden of multiple overlapping root causes. These autoantibodies are not random findings. They are the signature of loss of immune tolerance — the point at which the immune system can no longer distinguish self from non-self in neural tissue. And that loss of tolerance, more often than not, begins far from the brain.
This article walks through the major drivers of neuroinflammation I encounter in practice, how they converge to break immune tolerance, and why restoring immune regulation — not simply suppressing inflammation — is the key to recovery.
But first, a clinical note: when I say neuroinflammation, I am not describing a rare or exotic condition. Brain fog often reflects neuroinflammation. The client who cannot find their words, who walks into a room and forgets why, who feels like they are thinking through gauze — that is microglial activation, cytokine-mediated synaptic dysfunction, and impaired neurotransmitter metabolism. Headaches often involve neuroinflammation — whether tension-type, migraine, or the diffuse pressure that mold-ill clients so often describe. Neurogenic inflammation, mast cell degranulation in the meninges, and sensitized trigeminal pathways are all expressions of the same upstream processes. Anxiety and depression often involve neuroinflammation. The cytokine-driven activation of indoleamine 2,3-dioxygenase (IDO) shunts tryptophan away from serotonin synthesis and toward the kynurenine pathway, producing quinolinic acid — a potent NMDA receptor agonist that contributes to the very excitotoxicity we will discuss later. These are not always separate diagnoses. They are the lived experience of an inflamed brain, and many of the mechanisms in this article can be operating behind them.
The Gut: Where Tolerance Is Made and Lost
The gut houses roughly 70% of the immune system. More importantly, it is where immune tolerance is established and maintained. The gut-associated lymphoid tissue (GALT), Peyer’s patches, and the regulatory T-cell populations they support are responsible for teaching the immune system what to attack and what to leave alone. This is the primary site of barrier integrity and immune education in the body, and when it fails, everything downstream follows.
When intestinal barrier integrity is compromised — through dysbiosis, inflammatory foods, infections, or toxicant exposure — this education breaks down. Lipopolysaccharide (LPS) and other microbial components translocate into systemic circulation, triggering toll-like receptor 4 (TLR4) activation on immune cells throughout the body. But the deeper problem is that loss of intestinal barrier integrity disrupts peripheral immune tolerance — the regulatory T-cell function, dendritic cell tolerogenicity, and mucosal immune calibration that keep the immune system from attacking self-tissue. This is not simply about reacting to food antigens. When gut barrier function fails, the constant antigenic bombardment exhausts regulatory mechanisms system-wide, lowering the threshold for autoimmune reactivity everywhere. Proteins that were previously tolerated — including self-proteins in distant tissues like the brain — become targets.
The vagus nerve provides a direct neural highway from gut to brainstem. Inflammatory signaling in the gut alters vagal afferent tone, which directly modulates central neurotransmitter systems, microglial activity, and hypothalamic function. Simultaneously, short-chain fatty acids (SCFAs) produced by commensal bacteria — particularly butyrate — are critical for maintaining blood-brain barrier integrity. When gut dysbiosis reduces SCFA production, the blood-brain barrier becomes more permeable, allowing peripheral inflammatory mediators and autoantibodies direct access to neural tissue.
This is the “leaky gut to leaky brain” progression. It is not merely a pathway to inflammation — it is a pathway to autoimmunity.
The Oral Microbiome: An Amplifier That Can Become a Primary Driver
In most clients, gut barrier disruption is the primary event — but the oral microbiome is an underappreciated amplifier that can also become a primary driver under certain conditions. The oral cavity is not a separate ecosystem; it is directly connected to the gut through the simplest physiology imaginable: saliva. Most adults produce roughly 0.5-1.5 liters per day, and with it swallow on the order of 10^11 to 10^12 oral bacteria daily. When gut dysbiosis and systemic inflammation alter immune function, the oral microbiome shifts in parallel — and oral dysbiosis then feeds pathogenic organisms and inflammatory mediators right back into the gut, creating a bidirectional cycle.
Where the oral cavity becomes a primary driver rather than an amplifier is in clients with direct oral insults — most notably mercury amalgam fillings, root canal infections, and cavitations. Mercury vapor from amalgams suppresses local immune function, disrupts the oral microbiome, and provides a constant source of toxic metal exposure that drains directly into the lymphatic and venous systems of the head and neck. In these clients, the oral cavity is not downstream of the gut — it is an independent source of immune dysregulation and toxic burden that compounds whatever is happening below.
Porphyromonas gingivalis, the keystone pathogen in periodontal disease, illustrates why oral health cannot be ignored regardless of the direction of causality. Researchers have identified P. gingivalis and its toxic proteases called gingipains directly in the brains of Alzheimer's patients. Published work by Dominy et al. (2019) in Science Advances demonstrated gingipain presence in postmortem Alzheimer’s brain tissue and showed that gingipain inhibitors reduced bacterial load and neurodegeneration in animal models. This organism does not merely correlate with neurodegeneration — it may actively drive it through protease-mediated neuronal damage and chronic immune activation.
The gut-brain axis is really a mouth-gut-brain axis. Whether oral dysbiosis is cause or consequence in a given client, it must be assessed and addressed — because left unchecked, it perpetuates the very gut and systemic inflammation that drives neuroinflammation.
Blood-Brain Barrier Permeability: The Convergence Point
Nearly every insult discussed in this article damages the blood-brain barrier (BBB). The BBB depends on tight junction proteins — claudin-5, occludin, ZO-1 — maintained by astrocytic end-feet and pericytes. These structures are vulnerable to systemic inflammation, oxidative stress, mast cell activation, heavy metals, mycotoxins, and infectious agents.
Once BBB integrity is lost, the brain shifts from tightly regulated immune surveillance into pathological access. Peripheral cytokines, autoantibodies, toxicants, and pathogens gain access to neural tissue. And critically, neuronal and glial antigens that are normally sequestered behind the BBB become visible to the peripheral immune system. This is how neuronal damage leads to autoimmune targeting: when myelin basic protein (MBP), gangliosides, synapsin, tubulin, and other neural components leak through a compromised barrier, the already-primed immune system recognizes them as foreign.
This is one plausible mechanism behind demyelinating patterns and autoimmune-targeting of myelin. Autoantibody panels are not diagnostic of multiple sclerosis (MS), but they can be a clue that immune tolerance has been lost and myelin is under immune pressure—especially in the right clinical context. When a client develops numbness, tingling, visual changes, or coordination difficulties, it raises the question of demyelination and warrants a formal neurologic workup; in parallel, I investigate why the barrier broke and why tolerance was lost.
Addressing the BBB is therefore rarely optional in complex cases. It is central to any neuroinflammation protocol, and its integrity depends on immune regulation, gut health, and the resolution of the upstream insults that damaged it.
It is also worth noting that certain brain structures never had full BBB protection to begin with. The circumventricular organs (CVOs) — including the area postrema, the subfornical organ, the organum vasculosum of the lamina terminalis, the median eminence, and the pineal gland — are regions where the BBB is fenestrated or absent by design, allowing them to sample blood-borne signals for neuroendocrine regulation. This means that systemic toxicants, heavy metals, and inflammatory mediators have direct access to these structures without requiring BBB breakdown. The clinical implications are significant: the area postrema’s exposure explains the nausea so common in toxic clients; the median eminence’s vulnerability affects hypothalamic-pituitary signaling; and the pineal gland’s direct exposure to circulating metals and fluoride contributes to its well-documented calcification with age — a process that impairs melatonin production and further disrupts the circadian regulation and antioxidant protection that the brain depends on.
Stealth Infections: When the Immune System Cannot Keep Up
Chronic toxic and biotoxic exposures suppress innate and adaptive immune function in ways that open the door to opportunistic and stealth infections. Organisms like Borrelia and Bartonella species have well-documented neurotropism. Borrelia can cross the BBB, persist intracellularly, form biofilms, and directly infect glial cells and neurons. Bartonella has been identified in brain tissue and is increasingly recognized as a cause of neuropsychiatric symptoms including encephalopathy, seizures, and mood instability. Rage/anger and sudden explosive irritability are particularly characteristic of Bartonella neuro-invasion — a clinical pattern distinct enough that its presence should prompt investigation for this organism, especially in children.
These organisms physically inhabit neural tissue, triggering both direct cytotoxicity and secondary autoimmune responses. The molecular mimicry between microbial surface proteins and human neural antigens is well-documented — Borrelia outer surface proteins share epitopes with myelin and neuronal gangliosides. The immune response intended for the pathogen cross-reacts with self-tissue, and in a client who has already lost tolerance, this cross-reactivity is amplified rather than contained.
When the immune system is already compromised — by mold exposure, heavy metals, or chronic stress — these infections establish themselves and perpetuate a cycle of immune activation and neurological damage that is extremely difficult to interrupt without addressing all layers simultaneously.
Viral Reactivation: A Consequence of Terrain, Not a Primary Cause
A discussion of stealth infections would be incomplete without addressing the role of latent viruses — Epstein-Barr virus (EBV), human herpesvirus 6 (HHV-6), cytomegalovirus (CMV), and others. These viruses are ubiquitous; the vast majority of the adult population carries them. And in a well-regulated immune system, they remain dormant and clinically irrelevant. This is a critical point: viruses do not typically cause problems when immune function is intact. We see viral reactivation become clinically significant precisely because the platform is already imbalanced and the terrain is dysfunctional.
When mold exposure, heavy metals, chronic infections, or other toxic burdens suppress NK cell function, T-cell surveillance, and interferon signaling, latent viruses escape immune containment and reactivate. HHV-6 is directly neurotropic — it has been identified in multiple sclerosis plaques and temporal lobe epilepsy tissue. The landmark Bjornevik et al. (2022) study published in Science demonstrated a 32-fold increased risk of MS following EBV infection, strongly implicating EBV in the autoimmune demyelination process through molecular mimicry between EBV nuclear antigen 1 (EBNA1) and the glial cell adhesion molecule GlialCAM.
But the clinical error is to treat viral reactivation as the root cause. In the clients I see, viral reactivation is a signal that the terrain has failed. It is the mold-driven immune dysregulation, the metal-mediated NK cell suppression, the cortisol dysregulation, and the barrier breakdown that allowed these viruses to emerge. Antiviral therapy may be a necessary component of treatment, but without restoring the immune regulation that should be keeping these viruses in check, reactivation will recur. The virus is the opportunist. The terrain is the cause.
Toxic Metals: Displacing What the Brain Needs Most
Toxic metals such as lead, mercury, cadmium, aluminum, and arsenic accumulate in neural tissue and exert damage through multiple mechanisms. One of the most clinically significant is their ability to displace zinc from metallothionein — the family of cysteine-rich proteins responsible for zinc and copper homeostasis, free radical quenching, and heavy metal detoxification.
When toxic metals occupy metallothionein binding sites, zinc becomes functionally unavailable. Zinc is a cofactor for over 300 enzymes including superoxide dismutase, alkaline phosphatase, and δ-aminolevulinic acid dehydratase (involved in heme synthesis). It is essential for immune regulation, neurotransmitter metabolism, and synaptic plasticity. Its displacement creates a cascade of dysfunction that extends far beyond simple “toxicity.”
Mercury also collapses antioxidant capacity by targeting thiols. Mercury has a strong affinity for sulfur-containing thiol groups, and in practice this means it binds and depletes glutathione — the primary intracellular antioxidant that protects neurons from oxidative stress. When glutathione drops, reactive oxygen species rise, lipid membranes become vulnerable to peroxidation, and mitochondria become easier to derail. This is one reason metal burden so often shows up as “brain inflammation” rather than just a vague toxicity story.
Lead is neurotoxic in a different way than mercury — it behaves like calcium. Lead can substitute for calcium in voltage-gated calcium channels and calcium-binding proteins, disrupting synaptic signaling and neurotransmitter release. This is one reason lead exposure is so strongly associated with cognitive impairment: it doesn’t just “poison” neurons, it scrambles the electrical language they use to communicate. Lead also amplifies oxidative stress and mitochondrial dysfunction, pushing neural tissue toward excitotoxicity and inflammatory signaling. And mechanistically, it hits the heme pathway hard: lead inhibits enzymes like δ-aminolevulinic acid dehydratase and ferrochelatase, reducing heme synthesis and further stressing mitochondria. In a brain already carrying inflammatory burden, lead becomes a multiplier — destabilizing signaling, weakening energy production, and lowering the threshold for neuroinflammation.
Aluminum deserves its own mention because its neurotoxicity is mechanistically plausible even when exposure is chronic and low-grade. Aluminum has been shown in experimental models to drive oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, and glial activation — the same core ingredients that turn a stressed brain into an inflamed brain. It also interacts with protein aggregation pathways relevant to neurodegeneration, including amyloid-β and tau, and there are human studies linking higher environmental aluminum exposure (particularly via water and occupational settings) with increased dementia risk — though the evidence is mixed and not uniformly consistent across studies. One detail that is especially relevant mechanistically: aluminum citrate appears capable of transport across the blood–brain barrier via amino-acid transport systems, tying aluminum exposure to redox balance and glutathione-dependent defenses. Notably, aluminum also can accumulate in the pineal gland — compounding the pineal calcification and melatonin decline we will be discussing in this article. In a system already burdened by inflammation, impaired detoxification capacity, and mitochondrial strain, aluminum can behave like a multiplier rather than a single-cause villain.
Cadmium is a uniquely insidious toxic metal because it accumulates slowly and binds tightly to metallothionein, effectively hijacking the body’s metal-handling and antioxidant infrastructure. It depletes glutathione indirectly by driving oxidative stress and consuming redox capacity, and it disrupts the function of zinc- and selenium-dependent enzymes that neurons rely on to stay protected from reactive oxygen species. Cadmium also impairs mitochondrial function—reducing ATP production while increasing oxidative damage—pushing cells toward the same danger signaling and inflammatory loops described throughout this article. Over time, cadmium contributes to mineral dysregulation by displacing essential metals and altering signaling pathways that depend on calcium and zinc. Clinically, cadmium is common in smokers (tobacco is a major source), but it can also enter through food (especially certain shellfish, organ meats, rice, and cacao) and occupational exposures. In a system already burdened by inflammation and impaired detoxification, cadmium acts less like a single trigger and more like a chronic amplifier—lowering resilience, stressing mitochondria, and making neuroinflammation easier to sustain.
Neuromelanin and Metal-Induced Cell Death
Neuromelanin, the dark pigment found in dopaminergic neurons of the substantia nigra and noradrenergic neurons of the locus coeruleus, normally serves a protective role by chelating excess iron, copper, and other transition metals. However, when metal burden exceeds neuromelanin’s binding capacity, free metals catalyze Fenton chemistry — generating hydroxyl radicals that overwhelm antioxidant defenses and trigger lipid peroxidation, mitochondrial membrane damage, and ultimately neuronal death.
This mechanism is directly implicated in the pathogenesis of Parkinson’s disease. Elevated iron in the substantia nigra, combined with reduced ferritin and impaired ceruloplasmin function, creates an environment where neuromelanin-containing neurons are selectively destroyed. The same principle applies more broadly: any condition that increases brain metal burden while impairing metal-binding and antioxidant capacity accelerates neurodegeneration.
Pesticides, Paraquat, and the Parkinson’s Connection
Heavy metals are not the only environmental toxicants that target these vulnerable dopaminergic neurons. The herbicide paraquat is structurally similar to MPP+, the active metabolite of MPTP — the compound famously discovered to cause acute Parkinsonism in exposed individuals. Paraquat generates superoxide radicals through redox cycling, and epidemiological studies have consistently associated occupational and residential paraquat exposure with significantly increased Parkinson’s disease risk. The mechanism converges on the same endpoint: oxidative destruction of neuromelanin-containing neurons in the substantia nigra.
Beyond paraquat, the broader class of organochlorine and organophosphate pesticides contributes to neuroinflammation through mitochondrial complex I inhibition, glutathione depletion, and microglial activation. Glyphosate, the most widely used herbicide globally, warrants specific mention not only for its direct neurotoxic potential but for its impact on the gut — it disrupts tight junction integrity and interferes with the shikimate pathway in gut microbiota, impairing the microbial production of essential aromatic amino acids including tryptophan, tyrosine, and phenylalanine. These are the precursors to serotonin, dopamine, and norepinephrine. Glyphosate’s damage to the gut microbiome therefore feeds into the neurotransmitter deficiencies and gut barrier disruption discussed earlier in this article.
Copper, Mineral Dysregulation, and Neuroexcitotoxicity
Copper is both essential and potentially neurotoxic, and its homeostasis is tightly linked to inflammation. Pro-inflammatory states upregulate ceruloplasmin (an acute phase reactant that carries copper), while simultaneously impairing copper-zinc balance. Long term inflammatory states — particularly when accompanied by the cortisol resistance and HPA dysfunction described in an upcoming section — can also downregulate the production of ceruloplasmin, leading to copper trafficking dysfunction. The result is elevated unbound copper — a potent generator of reactive oxygen species and a direct contributor to excitotoxicity.
The excitotoxic cascade involves the NMDA receptor, calcium, magnesium, and glutamate. Under physiological conditions, magnesium occupies the NMDA receptor channel as a voltage-dependent plug, preventing excessive calcium influx. When magnesium is depleted — as it commonly is in chronically inflamed, stressed, or toxic clients — the NMDA receptor becomes overactive. Simultaneously, impaired astrocytic glutamate reuptake (via reduced expression of EAAT2 transporters, often driven by inflammation and oxidative stress) leads to synaptic glutamate accumulation.
The result is sustained NMDA receptor activation, excessive intracellular calcium influx, mitochondrial calcium overload, calpain activation, and neuronal death. This is classical glutamate excitotoxicity, and it is a core mechanism in conditions ranging from traumatic brain injury to chronic neuroinflammation. Calcium-magnesium dysregulation is therefore not a minor nutritional concern — it is a central node in the excitotoxic pathway.
EMF Exposure and Voltage-Gated Calcium Channels
The published work of Dr. Martin Pall has proposed a mechanism by which electromagnetic field (EMF) exposure contributes to calcium-mediated excitotoxicity through activation of voltage-gated calcium channels (VGCCs). Pall’s papers, published in journals including the Journal of Cellular and Molecular Medicine (2013) and Environmental Research (2018), argue that non-thermal EMF exposure activates VGCCs in cell membranes, leading to excessive intracellular calcium influx, downstream nitric oxide and peroxynitrite production, and oxidative stress.
This model is consistent with the known calcium-dependent excitotoxic pathways discussed above. While the body of literature on this topic continues to evolve and remains debated within mainstream toxicology, the published mechanistic work warrants clinical consideration — particularly in clients with high EMF exposure who present with neurological symptoms refractory to other interventions.
Biotoxins and the Olfactory Nerve: A Direct Route to the Brain
In water-damaged buildings, the problem is rarely one exposure — it’s a stack. Mold-derived mycotoxins (including trichothecenes, ochratoxin A, gliotoxin, and aflatoxins) matter not only because they drive systemic immune activation, but because inhaled exposures can also interface with the nervous system directly. Some compounds and inflammatory signals in the nasal cavity can reach the brain via the olfactory pathway, with access to the olfactory bulb and, potentially, deeper structures including limbic circuitry —bypassing the blood–brain barrier’s usual gatekeeping. Once neuroimmune signaling is active, the hypothalamus becomes a vulnerable target because it sits at the center of hormonal, autonomic, and circadian regulation.
For a deeper dive into mycotoxins, see: https://katalystforhealth.com/blog/f/happy-mold-vs-angry-mold-a-problem-of-mycotoxins-part-i
Mold can contribute mycotoxins, but it also sheds spores and fine fungal fragments that carry immune-activating cell-wall components like β-glucans, along with other fungal debris. Gram-negative bacteria contribute endotoxin (LPS), which strongly activates innate immune signaling. Actinobacteria (common in damp materials and dust) can add their own inflammatory burden through cell wall components and microbial metabolites. Clinically, the total “biotoxin load” is what drives the downstream loops I describe throughout this article — mast cell activation, barrier dysfunction, autonomic dysregulation, and microglial activation.
Alpha-Melanocyte Stimulating Hormone (MSH) Suppression
MSH is a neuropeptide with broad anti-inflammatory and mucosal regulatory functions. It modulates cytokine production, supports mucosal barrier integrity throughout the gut and respiratory tract, has antimicrobial properties, and regulates melanocortin pathways involved in appetite, energy balance, and immune tolerance. When hypothalamic damage suppresses MSH production — as is often seen in chronically mold-exposed clients — the downstream effects are profound: increased mucosal permeability, loss of immune regulation at barrier surfaces, heightened susceptibility to colonization by MARCoNS (multiple antibiotic resistant coagulase negative staphylococci) and other opportunistic organisms, and a self-perpetuating inflammatory state that resists treatment until MSH status is addressed.
Low MSH is one of the reasons mold-ill clients remain symptomatic long after exposure has ended. Without adequate MSH, mucosal barriers cannot heal, immune regulation at every barrier surface is impaired, and the ongoing antigenic exposure perpetuates loss of tolerance. MSH is not merely a marker of hypothalamic injury — it is a functional gatekeeper of the immune tolerance that prevents autoimmunity.
Orexin Suppression and Circadian Disruption
Orexin (hypocretin), produced by a small population of hypothalamic neurons, is essential for maintaining wakefulness, regulating the cortisol awakening response (CAR), and coordinating the circadian clocks of peripheral tissues. When mycotoxin-mediated hypothalamic injury reduces orexin output, the consequences extend far beyond sleepiness.
The cortisol awakening response — the sharp rise in cortisol within the first 30–45 minutes after waking — is an orexin-dependent process that sets the circadian rhythm for virtually every cell in the body. When it is blunted, downstream clocks desynchronize, affecting metabolism, immune cycling, hormone secretion, and autonomic tone.
Low orexin also contributes to symptoms consistent with postural orthostatic tachycardia syndrome (POTS). Orexin neurons project to sympathetic preganglionic neurons and the rostral ventrolateral medulla, and their loss impairs the autonomic adjustments required for postural changes. Many clients with biotoxin illness who present with orthostatic intolerance are experiencing orexin-mediated autonomic failure that will not respond to conventional POTS treatments alone.
HPA Axis Dysregulation and Cortisol Resistance
The hypothalamic-pituitary-adrenal (HPA) axis is not merely suppressed in chronically ill clients — it is dysregulated in a way that creates cortisol resistance. Under normal conditions, cortisol is the body’s primary endogenous anti-inflammatory signal: it activates glucocorticoid receptors on immune cells to suppress NF-κB-mediated cytokine production. However, chronic inflammation creates a paradox. Sustained NF-κB activation downregulates glucocorticoid receptor expression and sensitivity, meaning that even when cortisol is present, immune cells stop responding to it.
This is cortisol resistance — and it explains the “wired and tired” presentation so common in these clients. The HPA axis may be producing cortisol (or even overproducing it in early stages), but the anti-inflammatory signal is not being received. The immune system operates as though cortisol is absent, perpetuating inflammation despite what appears to be adequate adrenal output on standard testing. Over time, as mycotoxin-mediated hypothalamic damage compounds the picture and orexin-dependent CAR blunts, the HPA axis itself begins to fail — and the client transitions from cortisol resistance to cortisol insufficiency, losing both the signal and the response.
PANS: A Window into Immune-Mediated Neuroinflammation
Pediatric Acute-onset Neuropsychiatric Syndrome (PANS) offers one of the clearest clinical illustrations of how immune dysregulation translates directly into neuroinflammation — and working with this population has deeply informed my understanding of these mechanisms in clients of all ages.
PANS is characterized by the sudden onset of obsessive-compulsive symptoms, tics, anxiety, emotional lability, cognitive regression, and behavioral changes. The mechanism involves autoantibodies targeting the basal ganglia — the brain region responsible for motor control, habit formation, and executive function. Conventional models attribute PANS to post-infectious molecular mimicry, most commonly following streptococcal infection. And while infection is often the proximal trigger, what I see in practice tells a more layered story. Notably, when rage is a prominent symptom in a PANS child — the sudden, explosive, seemingly unprovoked anger that is so distressing to families — investigating Bartonella is essential.
In my caseload, mold exposure shows up so consistently that I treat it as a primary platform until proven otherwise. It is the mycotoxin-driven immune dysregulation — the suppression of MSH, the impairment of regulatory T-cell function, the disruption of mucosal barriers, and the resulting chronic immune activation — that creates the terrain on which any subsequent trigger can push the system into frank autoimmunity. The streptococcal infection, the viral illness, the vaccine, the environmental exposure — these are the match, but mold built the kindling.
This is why treating PANS with antibiotics or IVIG alone, without addressing the underlying mold exposure and the immune dysregulation it creates, so often results in relapsing-remitting courses. The basal ganglia autoantibodies are a downstream expression of a system that has fundamentally lost its ability to regulate immune responses. Until that regulatory capacity is restored — by removing the biotoxic burden, healing barriers, balancing minerals and re-establishing tolerance — the autoimmunity will find new targets with each new trigger.
The PANS model is instructive because it makes visible what is happening more subtly in many adult clients with neuroinflammation: a toxic platform, usually mold-driven, at least that is what I see in my practice, creates systemic immune dysregulation, and then relatively minor triggers produce disproportionate neuroimmune responses. The autoantibodies to basal ganglia tissue in PANS are the same phenomenon as the autoantibodies to MBP, synapsin, gangliosides, and dopamine receptors I see on autoantibody testing against nervous system tissue in adults — different targets, same underlying loss of tolerance.
Traumatic Brain Injury: The Priming Event
One factor that is frequently overlooked in the neuroinflammation history is remote traumatic brain injury (TBI) — including concussions that were considered mild or were never formally diagnosed. Even a single mild TBI primes microglia into a “sensitized” state. These microglia do not return fully to their surveilling phenotype. Instead, they remain in a para-inflammatory state, morphologically and functionally altered, with a lower activation threshold for subsequent insults.
This means that a client who sustained a concussion years or even decades before a mold exposure, a Lyme infection, or a toxic metal accumulation will mount a more aggressive and more sustained neuroinflammatory response to those insults than a client without that priming event. The TBI itself may have been clinically resolved — but the microglial landscape it left behind was not. Repetitive mild TBI, as seen in contact sports, domestic violence, and military service, creates cumulative priming that further lowers the threshold.
When a neuroinflammatory presentation seems disproportionate to the current burden — when the mold exposure was modest, the metal levels are not extreme, the infections are manageable, but the neurological symptoms are severe — a TBI history is often the missing variable. It does not change the treatment approach fundamentally, but it explains why the brain is responding the way it is and sets appropriate expectations for the pace of recovery.
COVID-19: The Straw That Broke Immune Tolerance
No discussion of neuroinflammation in the current clinical landscape is complete without addressing COVID-19. SARS-CoV-2 has demonstrated neurotropic potential — the virus can interface with the nervous system via the olfactory pathway, via hematogenous spread across a compromised blood–brain barrier, and through infection or dysfunction of brain endothelial cells and pericytes. Published research has documented microglial activation, astrocyte reactivity, BBB disruption, and neuronal injury in COVID-19 patients, including those with mild acute illness. Autopsy studies have shown inflammatory changes in regions including the brainstem, hippocampus, and cortex.
But the more clinically relevant question for many clients is not acute COVID — it is Long COVID. In my view, Long COVID often behaves like a vascular–immune lock-in loop in at least a subset of patients. Multiple groups have reported abnormal fibrin(ogen)-rich “microclots” (sometimes described as amyloid/fibrinaloid fibrin), platelet activation, and impaired fibrinolysis. The proposed consequence is microvascular flow limitation — tiny regions of tissue hypoxia that can sustain fatigue, exertional intolerance, dysautonomia, and cognitive impairment, while also perpetuating inflammatory signaling. This remains an active area of research and debate, but mechanistically it fits the same physiology described in the hypercoagulation section of this article: inflammation drives clotting, clotting worsens oxygen delivery, and hypoxia feeds inflammatory signaling.
At the same time, Long COVID increasingly looks like a persistence-and-immune-activation problem in some cases — particularly in the gut. Tissue studies and emerging reviews describe SARS-CoV-2 antigen persistence in the gastrointestinal tract months after infection, with ongoing immune activation. Notably, double-stranded SARS-CoV-2 RNA has been detected in gut tissue in some participants — a finding consistent with ongoing viral RNA biology and potentially replication activity within tissue immune compartments. If antigenic material persists in gut immune tissue and keeps innate and adaptive immune cells chronically activated, that persistent cytokine and complement tone becomes a plausible upstream driver of vascular dysfunction, BBB permeability, and microglial activation — i.e., chronic neuroinflammation without an ongoing acute infection.
Another persistence model involves the immune cells themselves. Patterson and colleagues reported SARS-CoV-2 S1 protein within CD16+ monocyte subsets in individuals with Long COVID months after infection, alongside immune dysregulation. Their work suggests that even without clear evidence of full-length replicating virus in the bloodstream, antigenic remnants inside long-lived or recirculating monocytes could act as a mobile inflammatory stimulus — interacting with endothelium, platelets, and cytokine networks in a way that sustains vascular inflammation and neuroimmune activation. This is not the only explanation for Long COVID, and causality is still being worked out, but the mechanism is coherent: chronically activated innate immune cells plus vascular dysfunction can keep microglia primed and the brain inflamed long after the acute infection resolves.
What I observe clinically fits this broader systems model. I have had clients come to me with aphasia and significant memory loss in their early 40s — presentations that would be alarming at any age, let alone decades away from typical neurodegenerative timelines. In these cases, COVID was the proximal trigger — but rarely the root cause. It was the straw that broke an immune system whose tolerance was already compromised by pre-existing burdens — mold exposure, accumulated metals, chronic gut dysfunction, metabolic stress — that had not yet produced obvious neurological symptoms.
COVID’s neuroinflammatory mechanisms — endothelial injury and ACE2-related vascular effects, complement activation, molecular mimicry between viral epitopes and human antigens, and direct microglial activation via innate immune signaling — are significant in their own right. But they are amplified enormously in a system that has already lost regulatory capacity. The virus exploits the same BBB vulnerability, impaired vagal tone, mast cell instability, and loss of immune tolerance that every other insult in this article can create. This is why some people recover uneventfully while others develop devastating, persistent neurological sequelae. The difference is the terrain.
In other words, COVID can be both an insult and an amplifier — but whether it becomes a long-term neuroinflammatory state depends on what it lands on.
Microglial Activation: Where Immune and Nervous Systems Become One
Microglia are the resident immune cells of the central nervous system, and their activation is both a consequence of and contributor to neuroinflammation. In their surveilling state, microglia monitor the neural environment and support synaptic pruning and plasticity. When activated by any of the insults described above — infections, toxins, metals, metabolic dysfunction — they shift to a pro-inflammatory phenotype, releasing TNF-α, IL-1β, IL-6, reactive oxygen species, and glutamate.
This creates a bidirectional amplification loop: systemic inflammation activates microglia, microglial activation generates neuroinflammation, neuroinflammation impairs neural control of immune function, and impaired neural-immune regulation worsens systemic inflammation. This is why neuroinflammation, once established, tends to be self-perpetuating.
Complement is one of the brain’s most underappreciated “wiring” mechanisms. Complement proteins don’t just fight infections — in the brain, they can act like molecular tags that mark synapses. Microglia can recognize those tags and prune synapses as part of normal development and repair. But when the system is chronically inflamed, that same machinery can become miscalibrated — and the brain starts losing connections it can’t afford to lose. This is one of the cleanest bridges between immune activation and the lived experience of cognitive slowing, mood shifts, sensory overwhelm, and “I don’t feel like myself.”
The Vagus Nerve and the Cholinergic Anti-Inflammatory Pathway
The vagus nerve is the primary conduit of the cholinergic anti-inflammatory pathway. Vagal efferent signaling releases acetylcholine (ACh), which acts on alpha-7 nicotinic acetylcholine receptors (α7nAChR) on macrophages and other immune cells to suppress NF-κB-mediated cytokine production. This is the body’s endogenous brake on systemic inflammation.
When vagal tone is impaired — by gut inflammation, biotoxin injury, autonomic dysfunction, or chronic stress — this brake fails. The result is unchecked peripheral and central inflammatory signaling. Clinically, impaired vagal tone manifests as reduced heart rate variability, poor digestive motility, heightened anxiety, and an exaggerated inflammatory response to stimuli that would normally be well-tolerated. Restoring vagal function is therefore not a “nice to have” — it is mechanistically necessary for resolving neuroinflammation.
Mast Cell Activation, Histamine, and Sleep Disruption
Mast cells are present in the brain (particularly in the thalamus, hypothalamus, and meninges) and throughout the gut. When systemic inflammation — driven by gut dysbiosis, infections, or toxicant exposure — activates mast cells, the consequences extend far beyond histamine. Mast cell activation syndrome (MCAS) involves the release of hundreds of mediators including prostaglandins, leukotrienes, tryptase, heparin, TNF-α, IL-6, and vascular endothelial growth factor (VEGF). These mediators collectively increase vascular permeability (including BBB permeability), amplify neuroinflammation, sensitize pain pathways, and dysregulate the autonomic nervous system.
MCAS is often the clinical bridge between biotoxin illness and neuroinflammation. Mycotoxins are potent mast cell activators, and in clients with mold exposure, MCAS frequently becomes self-perpetuating — the mast cells remain in a hyperreactive state long after the initial trigger, responding to foods, chemicals, temperature changes, and stress with disproportionate mediator release. This is why so many clients with complex neuroinflammatory presentations also carry a pattern of multi-system reactivity that looks like “allergies to everything.” It is not allergy. It is a destabilized mast cell population that has lost its regulatory constraints.
Sex hormones are one of the most under-recognized mast cell amplifiers. Mast cells express hormone receptors, and estrogenic signaling can increase mast cell activation and mediator release — one reason histamine patterns and neuroinflammatory symptoms so often track with cycle changes, perimenopause, and estrogenic states. Progesterone, in contrast, has documented inhibitory effects on mast cell secretion in experimental models — which helps explain why a relative estrogen-to-progesterone imbalance can tip a sensitive client into worse histamine burden, more vascular permeability, and more neuroimmune noise. In a brain already struggling with sleep disruption and glymphatic congestion, that extra mast cell signal can be the difference between “tolerable” and “I’m on fire.”
Histamine specifically warrants attention for its role in sleep disruption. Histamine is a wake-promoting neurotransmitter, and its elevation disrupts sleep architecture, particularly the ability to initiate and maintain deep slow-wave sleep and REM sleep. This is critically important because deep sleep is when the glymphatic system — an astrocyte-mediated, aquaporin-4-dependent clearance system — is most active. The glymphatic system removes metabolic waste from the brain, including amyloid-β and tau proteins.
Sleep is not just rest — it is clearance. During sleep, glymphatic flow ramps up and helps move metabolic waste and inflammatory byproducts out of brain tissue and into cerebrospinal fluid. From there, meningeal lymphatic vessels help drain fluid and immune traffic out of the skull toward the deep cervical lymph nodes. When sleep is disrupted (or the system is congested), clearance slows — and inflammatory signaling has more time to echo.
When histamine-driven sleep disruption impairs glymphatic clearance, neurotoxic waste accumulates, further activating microglia and perpetuating neuroinflammation. This creates yet another self-reinforcing loop: inflammation → mast cell activation → histamine → poor sleep → impaired glymphatic clearance → more neuroinflammation.
Melatonin sits at the center of this vulnerability. The work of Dr. Russell Reiter — spanning decades and hundreds of publications — has established that melatonin is far more than a sleep-onset signal. It is one of the most potent endogenous antioxidants the body produces, and critically, it is synthesized within mitochondria themselves, where it scavenges hydroxyl radicals, peroxynitrite, and singlet oxygen at the very site of greatest oxidative stress. It upregulates glutathione peroxidase, superoxide dismutase, and catalase — the same antioxidant defenses that every insult in this article depletes. Melatonin production declines markedly with age, and that decline has been associated across many neurodegenerative conditions: Alzheimer's, Parkinson's, ALS, multiple sclerosis, Huntington's, and poor outcomes following stroke and TBI.
As melatonin falls, the brain loses both the circadian signal that initiates the deep sleep required for glymphatic clearance and the mitochondrial antioxidant protection that should be operating during that sleep. The pineal calcification discussed earlier — driven by the same metals, fluoride, and chronic inflammation described throughout this article — accelerates this decline far beyond normal aging. The result is a brain that cannot clear its waste, cannot protect its mitochondria, and cannot achieve the restorative sleep states it needs to resolve inflammation. Melatonin loss is not a minor age-related inconvenience — it is a mechanistic accelerant of every neurodegenerative process discussed in this article.
Hypercoagulation and Neuronal Hypoxia: The Silent Starve
Inflammation is inherently procoagulant. This is by evolutionary design — the coagulation cascade and the inflammatory cascade share common mediators and were originally part of the same host defense system. But in chronic neuroinflammation, this relationship becomes pathological.
Pro-inflammatory cytokines — particularly IL-6, TNF-α, and IL-1β — upregulate tissue factor expression on monocytes and endothelial cells, activate platelets, increase fibrinogen production, suppress natural anticoagulant pathways (protein C, protein S, antithrombin III), and impair fibrinolysis by elevating plasminogen activator inhibitor-1 (PAI-1). The net result is a hypercoagulable state characterized by increased fibrin deposition in the microvasculature — not necessarily large-vessel clotting, but a diffuse microvascular coagulopathy that impairs blood flow at the capillary level.
In the brain, this microvascular coagulopathy translates directly to neuronal hypoxia. Neurons are exquisitely oxygen-dependent — the brain consumes roughly 20% of the body’s oxygen despite comprising only 2% of its mass. Under normal conditions, the astrocyte-neuron lactate shuttle is a vital partnership: astrocytes take up glucose, convert it to lactate via aerobic glycolysis, and shuttle it to neurons via monocarboxylate transporters (MCT1 and MCT4 on astrocytes, MCT2 on neurons). Neurons then convert lactate to pyruvate and feed it into mitochondrial oxidative phosphorylation — an elegant system that provides neurons with a preferred fuel source. However, in hypoxic conditions, this partnership breaks down. Neurons can no longer perform oxidative phosphorylation efficiently, so the lactate they receive cannot be metabolized. Simultaneously, astrocytes upregulate glycolysis in response to hypoxia, producing even more lactate. The result is lactate accumulation that overwhelms the astrocytic buffering capacity, drives local acidosis, impairs pH-sensitive enzymatic function, and paradoxically inhibits the very astrocytic glutamate reuptake (via EAAT2) that prevents excitotoxicity. The neuron is simultaneously starved of usable energy and drowning in a substrate it can no longer process.
This is one of the underappreciated mechanisms behind brain fog. The client’s neurons are not just inflamed — they are hypoxic. And the hypoxia itself becomes another driver of inflammation, because HIF-1α (hypoxia-inducible factor) activation promotes NF-κB signaling, mast cell degranulation, and VEGF-mediated vascular leak, further compromising BBB integrity. Fibrin itself is directly neurotoxic — it activates microglia via CD11b/CD18 receptors and promotes demyelination, linking the coagulation cascade directly to the autoimmune targeting of myelin discussed earlier. In Alzheimer’s disease, the fibrin connection goes even deeper: fibrinogen interacts directly with amyloid-β, forming fibrin-amyloid complexes that are resistant to degradation and that promote sustained microglial activation and vascular inflammation. Research by Bhatt et al. and others has demonstrated that these abnormal fibrin deposits co-localize with amyloid plaques, and that disrupting the fibrinogen-amyloid-β interaction reduces neuroinflammation and cognitive decline in animal models. Hypercoagulation is not a peripheral finding in neurodegeneration — it is woven into the pathology itself.
Clinically, hypercoagulation is often invisible on standard labs. A CBC and basic metabolic panel will not reveal it. It requires specific assessment — D-dimer, fibrinogen, thrombin-antithrombin complexes, PAI-1, and in some cases functional platelet testing — to identify. When present, addressing the procoagulant state is essential for restoring oxygen delivery to neural tissue. Without it, every other intervention is working against a brain that is being slowly starved.
Insulin Resistance: Metabolic Fire in the Brain
Insulin resistance is not merely a metabolic condition — it is a neuroinflammatory one. And it is essential to understand that insulin resistance is not synonymous with type 1 or type 2 diabetes. It is a spectrum in which cellular glucose uptake and aerobic oxidative phosphorylation become progressively impaired — and by current estimates, the majority of the population exists somewhere on that spectrum. Any degree of insulin resistance means that cells, including neurons, are less efficient at producing ATP from glucose through their mitochondria. In the brain, which consumes roughly 25% of the body’s glucose supply, even modest impairments in insulin signaling have outsized consequences.
Chronic inflammation and toxicant exposure (particularly heavy metals and persistent organic pollutants) impair insulin receptor signaling in the brain. Cerebral insulin resistance is now so consistently associated with Alzheimer’s disease that many researchers refer to it as “type 3 diabetes.”
The mechanistic link centers on glycogen synthase kinase-3 beta (GSK-3β). Under normal conditions, insulin signaling inhibits GSK-3β via the PI3K/Akt pathway. When insulin resistance develops, GSK-3β becomes constitutively active, driving hyperphosphorylation of tau protein — the hallmark of neurofibrillary tangles — and promoting amyloid-β production and neuroinflammation.
Recent research on lithium is relevant here. Lithium is a potent GSK-3β inhibitor, and epidemiological data suggest that populations with higher lithium exposure through drinking water have lower rates of dementia. Clinical trials exploring low-dose lithium for neuroprotection are ongoing, and the GSK-3β mechanism provides a plausible explanation for lithium’s apparent cognitive benefits beyond its established role in mood stabilization.
Dr. Dale Bredesen’s work classifying Alzheimer’s subtypes has been valuable in this context. His Type 3 — toxic or inhalational Alzheimer’s — describes clients whose cognitive decline is driven primarily by biotoxin exposure, particularly mycotoxins. In my practice, I see this pattern frequently: clients presenting with progressive cognitive impairment, and when we investigate, mycotoxin burden is present alongside the metabolic and inflammatory markers that characterize neurodegeneration. The conventional model treats Alzheimer’s as a single amyloid-driven disease. The clinical reality is that many dementia presentations are the downstream expression of toxic exposures, immune dysregulation, and metabolic failure — and when those root causes are identified and addressed, cognitive trajectory can change in ways the conventional model does not predict.
Hormonal Dysregulation and Autoimmune Encephalopathy
Neuroinflammation does not occur in a hormonal vacuum. Two patterns are particularly significant in clinical practice:
Elevated prolactin from estrogenic states. Estrogen dominance — whether from impaired hepatic estrogen clearance, xenoestrogen exposure, or relative progesterone deficiency — stimulates lactotroph proliferation and prolactin secretion. Elevated prolactin is not benign neurologically. It is immunostimulatory: it promotes B-cell maturation, immunoglobulin production, and a Th2-skewed immune response that favors antibody-mediated autoimmunity. In the context of an already-inflamed brain with compromised barrier function, hyperprolactinemia drives the humoral immune system further toward autoantibody production — generating the very autoantibodies we detect on autoantibody testing targeting brain and nervous system tissue.
Low dopamine tone. Dopamine is a prolactin-inhibiting factor (via D2 receptors on lactotrophs), so dopamine deficiency and hyperprolactinemia are mechanistically linked. Dopamine synthesis depends on iron (as a cofactor for tyrosine hydroxylase), copper (for dopamine beta-hydroxylase), zinc, magnesium, and adequate methylation — all of which are disrupted in the toxic, inflamed clients described throughout this article. Furthermore, dopamine itself is anti-inflammatory within the CNS, acting on D1 and D2 receptors on microglia to suppress inflammatory cytokine production.
Low dopamine contributes directly to the anhedonia and depression so common in these clients — the inability to experience pleasure, the loss of motivation, the flatness that is often misattributed to psychological causes when it is fundamentally neurochemical. But the clinical picture is not simply “more dopamine is better.” Dopamine in excess is itself neurotoxic. Its auto-oxidation generates reactive quinones, hydrogen peroxide, and superoxide — the same oxidative chemistry that destroys neuromelanin-containing neurons in Parkinson’s disease. Elevated dopamine states, as observed in certain presentations of autism spectrum disorder, carry their own neurotoxic burden. This is consistent with findings of elevated homovanillic acid (HVA) in some autistic individuals, reflecting increased dopaminergic turnover and oxidative stress. The goal is rarely simply to raise or lower dopamine — it is to restore the regulatory balance that allows dopamine to function as a signaling molecule without becoming a source of damage. As with every system discussed in this article, the principle is the same: it is dysregulation, not simply deficiency or excess, that drives disease.
The convergence of elevated prolactin and low dopamine creates a neuroimmune environment highly permissive to autoimmune encephalopathy. On autoantibody testing, I often identify autoantibodies to myelin basic protein, gangliosides, tubulin, synapsin, dopamine receptors, NMDA receptors, and other neural targets. Each of these reflects a specific pattern of neural tissue damage and immune dysregulation — and each traces back to the layered insults described in this article. These are not idiopathic autoimmune findings. They are the predictable result of lost tolerance acting on a brain that has been damaged by toxins, infections, metals, and metabolic failure.
Membrane Integrity: Plasmalogens and Phosphatidylcholine
Every mechanism discussed in this article ultimately converges on the cell membrane. Neuronal membranes are extraordinarily rich in specialized lipids — particularly plasmalogens and phosphatidylcholine — and their integrity determines whether neurons function, communicate, and survive.
Plasmalogens are a unique class of ether phospholipids that constitute up to 30% of phospholipids in the human brain. They serve as endogenous antioxidants (their vinyl-ether bond scavenges reactive oxygen species), structural components of synaptic membranes, and facilitators of membrane fusion events critical for neurotransmitter release. Plasmalogen levels are consistently reduced in Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis. When oxidative stress from metals, mycotoxins, and chronic inflammation depletes plasmalogens faster than they can be synthesized, membrane fluidity decreases, synaptic signaling deteriorates, and neurons become increasingly vulnerable to excitotoxic and immune-mediated damage.
Dietary factors compound this problem significantly. The modern Western diet is disproportionately high in polyunsaturated fatty acids (PUFAs), particularly omega-6 linoleic acid from seed oils and processed foods. While PUFAs are often marketed as “heart healthy,” their molecular structure — multiple double bonds susceptible to oxidation — makes them highly vulnerable to lipid peroxidation, especially in the pro-oxidant environment of an inflamed body. When PUFAs in cell membranes undergo peroxidation, they generate toxic aldehyde byproducts including 4-hydroxynonenal (4-HNE), malondialdehyde (MDA), and lysophosphatidylethanolamine (LPE). These are not minor metabolites. 4-HNE is a potent electrophile that forms adducts with proteins, DNA, and lipids, directly damaging mitochondrial electron transport chain complexes and inactivating critical enzymes. MDA crosslinks proteins and contributes to the formation of advanced lipid peroxidation end products. Together, these aldehydes are directly toxic to the mitochondrial inner membrane — where cardiolipin, the specialized phospholipid essential for electron transport chain function, resides. When cardiolipin is oxidized and damaged by lipid peroxidation products, mitochondrial efficiency collapses, ROS generation increases, and the cell shifts further toward the danger signaling described in the CDR model. A high-PUFA diet in a chronically inflamed client is not just unhelpful — it is actively providing the substrate for ongoing membrane and mitochondrial destruction.
Phosphatidylcholine (PC) is the most abundant phospholipid in cell membranes and is essential for membrane structure, cell signaling, and as a precursor for acetylcholine synthesis. Its production depends on adequate methylation capacity (via the PEMT pathway) and choline availability. In clients with impaired methylation, toxic burden, and chronic inflammation, PC synthesis is compromised — which simultaneously impairs membrane integrity and the cholinergic signaling that is critical for the vagal anti-inflammatory pathway discussed earlier.
Supporting membrane health through plasmalogen and phosphatidylcholine repletion is therefore not a peripheral intervention — it is addressing the structural foundation upon which every other neurological function depends. Without intact membranes, neurotransmitter signaling fails, mitochondria lose their membrane potential, and the BBB cannot maintain its tight junctions. Membrane health is cellular health.
When Inflammation Becomes a Cell-Death Program
When chronic inflammation stays on long enough, the question is not only “how inflamed is the brain?” — it becomes “what programs are getting activated inside cells?” There are several regulated cell-death pathways that can sit downstream of oxidative stress, immune activation, and metabolic collapse. These aren’t theoretical. They are the biochemical endpoints that turn dysfunction into degeneration.
Ferroptosis is one of the most relevant here: an iron-dependent form of regulated cell death driven by runaway lipid peroxidation — meaning membrane damage becomes the final common pathway. When oxidative stress, iron dysregulation, and fragile membranes stack together, ferroptosis becomes a very real candidate mechanism for decline, not just symptoms.
Necroptosis is another: a programmed form of inflammatory cell death that can be triggered by innate immune signaling and cytokine pathways, and it tends to amplify inflammation because it ends in membrane rupture and danger signaling. Cuproptosis is emerging research, but clinically interesting given how often mitochondrial stress shows up in complex cases: copper inside cells can trigger a distinct regulated death pathway tied to mitochondrial metabolism. I mention these not to overwhelm, but to make the point clear: when the biology is stuck in threat, the cell can eventually choose an “exit strategy.” Our job is to remove the threat before it gets there.
Mitochondrial Dysfunction and the Cell Danger Response
Every insult discussed in this article damages mitochondria. Heavy metals inhibit electron transport chain complexes. Mycotoxins uncouple oxidative phosphorylation. Chronic infections increase metabolic demand while impairing energy production. Excitotoxic calcium influx collapses mitochondrial membrane potential. Hypercoagulation-driven hypoxia starves the very organelles that depend on oxygen to produce ATP.
When mitochondria fail, ATP production drops, reactive oxygen species generation increases, and the cell shifts toward glycolytic metabolism — which is itself pro-inflammatory. Damaged mitochondria release mitochondrial DNA (mtDNA) and other damage-associated molecular patterns (DAMPs) that activate innate immune receptors including the NLRP3 inflammasome and cGAS-STING, further amplifying inflammation. Mitochondrial dysfunction is therefore not merely a consequence of neuroinflammation — it is a driver.
Dr. Robert Naviaux’s Cell Danger Response (CDR) model provides an essential framework for understanding why mitochondrial dysfunction persists even after the initial threat has been removed. The CDR describes an evolutionarily conserved metabolic response in which cells detect danger — whether from infection, toxicants, physical trauma, or psychological stress — and shift mitochondria from their primary role of energy production to a defensive signaling role. Mitochondria begin releasing extracellular ATP — which, outside the cell, functions as a damage-associated molecular pattern (DAMP), one of the most potent danger signals in innate immunity. This extracellular ATP activates purinergic receptors (P2X7 in particular) on microglia, mast cells, and other immune cells, triggering NLRP3 inflammasome assembly and pro-inflammatory cytokine release. The mitochondria also release other purinergic signals that alter cellular metabolism and change gene expression to prioritize survival over normal function.
This is adaptive in the short term. But when the danger signals are chronic and layered — as they are in the clients described throughout this article, with ongoing mold exposure, persistent infections, accumulating metals, and unresolved gut dysfunction — the CDR does not resolve through its normal three stages. The cell remains locked in a defensive state. Mitochondria continue signaling danger rather than producing energy. The extracellular ATP that should resolve and allow healing instead perpetuates inflammation through P2X and P2Y purinergic receptor activation on immune and glial cells.
This is why so many chronically ill clients have the hallmarks of mitochondrial failure — profound fatigue, exercise intolerance, cognitive dysfunction, autonomic instability — without a primary genetic mitochondrial disease. Their mitochondria are not broken. They are responding to a persistent danger signal. The CDR model reframes the clinical challenge: it is not enough to support mitochondrial bioenergetics with cofactors and substrates, though that is necessary. The danger signals themselves must be resolved — the mold must be removed, the metals chelated, the infections addressed, the gut healed, the barriers restored — before mitochondria will shift back from defensive mode to energy production.
Mitochondrial membranes are themselves rich in cardiolipin and plasmalogens, linking mitochondrial health directly to the membrane integrity discussion above. When oxidative stress depletes these specialized lipids, mitochondrial electron transport becomes less efficient, generating more ROS in a vicious cycle.
Any comprehensive approach must address both the upstream triggers that activated the CDR and the bioenergetic support that mitochondria need to resume normal function once safety is restored.
Methylation: The Quiet Bottleneck
Methylation intersects with nearly every pathway discussed above. S-adenosylmethionine (SAMe) is required for neurotransmitter synthesis and degradation (COMT-mediated catecholamine metabolism), glutathione production (the primary intracellular antioxidant and detoxification molecule), phosphatidylcholine synthesis (cell membrane integrity), myelin maintenance, and epigenetic regulation of gene expression.
Polymorphisms in MTHFR, MTR, MTRR, BHMT, and CBS — combined with nutrient deficiencies in B12, folate, B6, and riboflavin — impair methylation capacity. In the context of heavy metal exposure (which depletes glutathione), chronic infection (which increases oxidative demand), and neuroinflammation (which requires robust neurotransmitter turnover), impaired methylation becomes a rate-limiting bottleneck that prevents recovery even when other interventions are in place.
One nuance that matters in stubborn neuroinflammatory cases: folate has to actually reach the brain. Folate receptor alpha (FRα) at the choroid plexus (where cerebrospinal fluid is made) transports active folate (5-MTHF) into the cerebrospinal fluid. In some clients, autoantibodies against FRα can interfere with that transport, creating a “cerebral folate deficiency” picture even when blood folate looks adequate. Clinically, this can show up as cognitive dysfunction, mood instability, headaches, seizures, or neurodevelopmental symptoms that look like neuroinflammation. In select cases, I consider folate receptor autoantibody testing (FRAT) and targeted support with folinic acid.
Immune Regulation: The Thread That Ties It All Together
If there is a single principle that unifies every section of this article, it is this: immune regulation is the master variable. Not immune suppression, not immune stimulation — immune regulation.
Androgen tone matters for immune regulation. Testosterone is not just about libido and muscle — it interfaces with immune signaling, and low testosterone commonly travels with a higher inflammatory burden. In a chronically stressed or toxic client, inflammation can suppress the gonadal axis, and the resulting drop in testosterone removes another layer of immune restraint. Clinically, this can show up as more cytokine-driven symptoms, more mast cell volatility, and less resilience to the same triggers that a better-regulated system would normally buffer.
Regulatory T cells (Tregs), tolerogenic dendritic cells, MSH-mediated melanocortin signaling, vagal cholinergic tone, androgen tone (testosterone), and adequate cortisol rhythms all participate in keeping the immune system calibrated. When any of these regulatory mechanisms are disrupted — by gut barrier breakdown, mycotoxin exposure, chronic infection, hormonal imbalance, or metabolic dysfunction — the immune system shifts from a state of tolerance to a state of reactivity. And once tolerance is lost, the immune system begins attacking the very tissues it is meant to protect.
Vitamin D signaling is another quiet regulator of immune tolerance. Immune cells can convert 25(OH)D into active 1,25(OH)2D locally, and that signaling tends to support a more regulatory immune tone. But in chronic inflammatory environments, the body can shift vitamin D metabolism — upregulating enzymes that inactivate active vitamin D (including 24-hydroxylase / CYP24A1) and downshifting receptor signaling in tissues. The result can be lower “bioactive” vitamin D signaling at the immune level, even when the serum numbers look deceptively fine. In a prolonged danger state — what Naviaux would frame as a CDR-like physiology — this is one more way tolerance can erode and autoimmunity can gain traction.
This is why the autoantibodies I see on testing panels are so diverse. They are not targeting one tissue for one reason. They are the expression of a fundamentally dysregulated immune system encountering neural antigens that have been exposed by barrier breakdown, cellular damage, and molecular mimicry. MBP antibodies, ganglioside antibodies, synapsin antibodies, dopamine receptor antibodies — each one tells us something about where the damage is, but the root cause is always the loss of immune tolerance that allowed the attack in the first place.
Restoring immune regulation means restoring the barriers (gut, BBB, mucosal) that prevent inappropriate antigen exposure. It means addressing the toxic burden (mold, metals, infections) that drives chronic immune activation. It means supporting the hormonal and metabolic milieu (MSH, cortisol rhythms, insulin sensitivity, dopamine) that the regulatory machinery depends on. And it means supporting the cellular and mitochondrial health that allows immune cells to function properly rather than defaulting to inflammatory programs.
There is no shortcut. But when we address these layers systematically, tolerance can be rebuilt, autoantibodies can diminish, and the brain can begin to heal.
Bringing It Together
Neuroinflammation is not one problem with one cause. It is the brain’s expression of total body burden — the sum of gut barrier breakdown and microbial imbalance, oral dysbiosis, toxic and biotoxic exposures, immune dysregulation, metabolic failure, hormonal disruption, membrane degradation, and impaired detoxification capacity. These systems do not operate in silos. They amplify each other through shared mechanisms: BBB breakdown, microglial activation, mitochondrial damage, excitotoxicity, loss of tolerance, and the erosion of neural-immune communication.
This is why single-target interventions so often fail these clients. Treating the infection without addressing the metals, clearing the mold without restoring the gut, supporting neurotransmitters without fixing mineral status and membrane integrity — these partial approaches cannot interrupt a cycle that operates across every system simultaneously.
The path forward requires identifying which layers are active in each individual and addressing them in the right sequence. Which tests matter depends on the case; testing should be guided by history, symptoms, and a clinician’s judgment. Gut, hormone, HTMA, organic acids, mycotoxin panels, heavy metal assessments, comprehensive metabolic panels, and advanced autoimmune testing and inflammatory markers — can provide the map. But understanding the interconnected biology is what allows us to read that map correctly.
The brain is rarely inflamed in isolation. To heal it, we must understand everything that is speaking to it.